Нейросети и физика: российские учёные повысили точность расчёта свойств молекул на 26%
Исследователи из Института органической химии РАН, Сколтеха, МГУ и ВШЭ разработали гибридный метод моделирования молекул, который использует нейросеть для тонкой настройки параметров, не нарушая физических законов. Новый подход позволяет предсказывать свойства соединений с высокой точностью, что критически важно для создания лекарств, катализаторов и материалов.
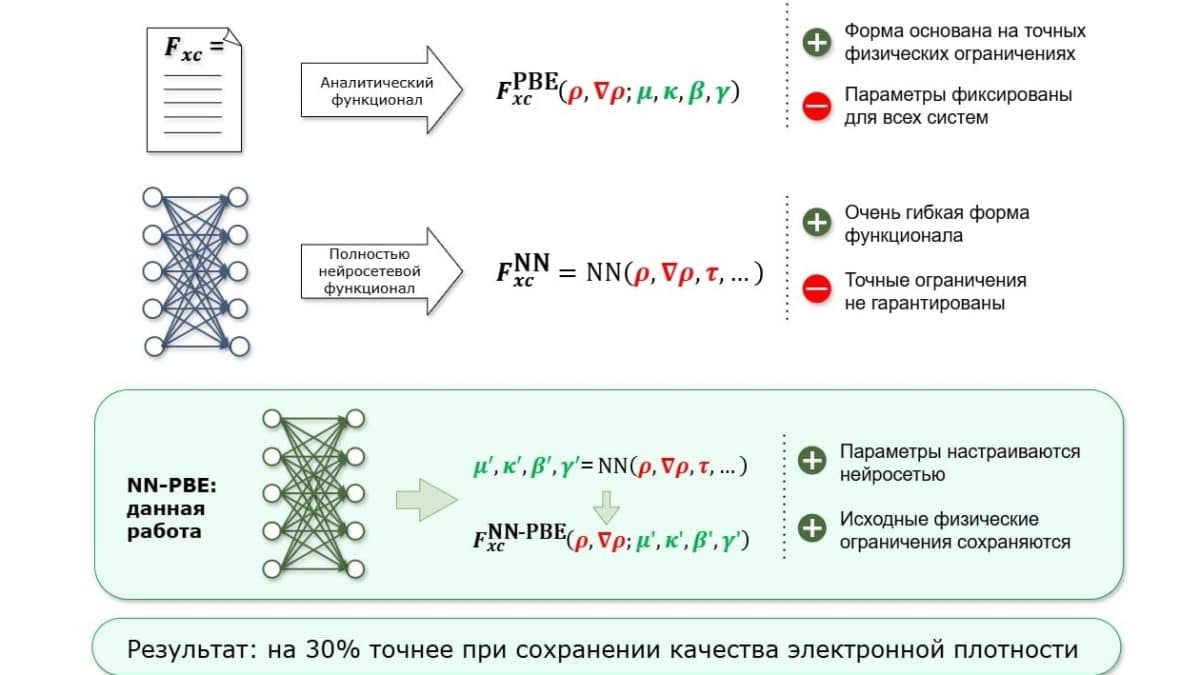
В современной вычислительной химии ключевым инструментом является теория функционала плотности (DFT). Она помогает понять, как электроны распределяются вокруг ядер молекулы, и на основе этого распределения рассчитать энергию, форму, прочность связей и реакционную способность. Однако точный универсальный функционал до сих пор не найден — учёные используют сотни моделей, каждая из которых имеет свои сильные и слабые стороны.
Два традиционных подхода и их ограничения
Существует два основных способа создания функционалов. Первый — строгое соблюдение известных физических законов, что даёт физически корректные, но не идеально точные результаты для самых разных соединений. Второй — эмпирический, когда гибкая математическая формула с множеством параметров подгоняется под известные экспериментальные данные. Такие функционалы точны на знакомых молекулах, но могут сильно ошибаться при расчёте новых веществ, игнорируя некоторые физические законы.
В последние годы появились нейросетевые функционалы, обучаемые на огромных массивах данных. Они демонстрируют впечатляющую точность, но часто «забывают» физику — нарушают фундаментальные ограничения, требуют много данных и плохо работают с незнакомыми системами. Учёным приходилось выбирать: либо точная физика и посредственная точность, либо высокая точность на известных соединениях и риск больших ошибок на новых.
Гибридный метод: лучшее из двух миров
Группа исследователей под руководством Михаила Медведева из ИОХ РАН предложила принципиально иной путь. Они взяли уже существующий функционал, который строго соблюдает ключевые физические законы, но имеет ограниченную точность расчёта энергий, и дополнили его нейросетью. Нейросеть обучена на примерах молекул с известными точными значениями энергий, чтобы самостоятельно выявлять скрытые закономерности и корректировать параметры функционала в зависимости от системы — при этом физические ограничения исходной модели остаются нерушимыми.
«Нам не пришлось отказываться от классических наработок, — комментирует Михаил Медведев. — Мы научили нейросеть локально подстраивать параметры уже существующего функционала, сохраняя ключевые заложенные в него создателями физические знания. Это как взять хорошо сконструированный двигатель и тонко его настроить, а не пытаться собрать новый из случайных деталей».
Новый инструмент протестировали на 30 типах химических реакций, последовательно уточняя электронную плотность до получения устойчивого результата. Оказалось, что усиленный нейросетью функционал проводит расчёты почти на 26% точнее, чем исходный функционал без нейросети. При этом гарантируется соблюдение физических законов — в отличие от чисто нейросетевых подходов, которые могут давать правильный ответ, нарушая физику.
Разработка открывает новые возможности для предсказания свойств соединений до проведения экспериментов. Такие расчёты востребованы при создании лекарств, катализаторов и новых материалов. В будущем учёные планируют объединить этот подход с ранее решённой проблемой «слепого пятна» в теории функционала плотности, чтобы создать ещё более надёжные функционалы для виртуального скрининга химических реакций и установления их механизмов.
Похожие статьи

Древнейшая вспышка чумы в Сибири: 5500-летние скелеты переписывают историю болезни
Учёные обнаружили в погребениях детей в Сибири древнейшие свидетельства вспышки чумы, датируемые 5500 лет. Это открытие отодвигает появление эпидемий чумы на тысячелетия назад.
Экспериментальная вакцина против фентанила показала многообещающие результаты на ранней стадии испытаний
Разработчик ARMR Sciences сообщил, что экспериментальная вакцина для предотвращения передозировок фентанилом продемонстрировала обнадеживающие результаты в ходе клинического испытания ранней фазы.

Тропический шторм «Артур» угрожает юго-востоку США сильными наводнениями
Первый в сезоне тропический шторм «Артур» обрушился на побережье Техаса и движется на юго-восток, неся с собой до 20 дюймов осадков и риск внезапных наводнений.
Комментарии
0 всего